Stereochemical Descriptors: Naming Molecules
Introduction
In my view the easiest way to describe the stereochemistry of a molecule is to draw it. But sometimes you will need to fully name a compound and this name will need to include information about stereochemistry. A method to define stereochemistry is also useful when manipulating molecules. It can help you check that you haven’t inadvertently changed a stereocentre.
The naming of stereoisomers has changed as our knowledge of the structure of molecules has improved. This post is primarily about the standardized stereochemical descriptors. But before I describe these descriptors, there are two older nomenclatures that are still in common use even though they have a number of draw backs.
The first is the use of + and – before a name, as in (–)-phenylalanine. This isn’t really a naming convention but a description of the physical properties of the molecule. The (+)-stereoisomer rotates plane polarised light clockwise and the (–)-enantiomer rotates light anticlockwise.
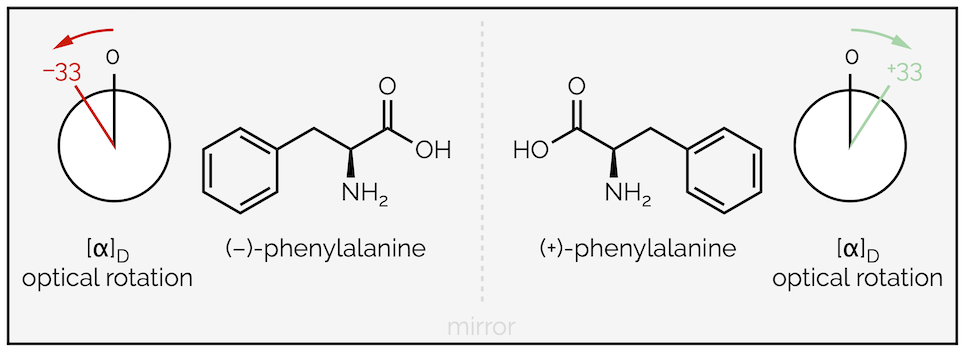
The use of (+) and (–) to describe the optical properties of stereoisomers
The second is the D or L, and if we keep with the amino acids, can be found in L-phenylalanine or even (L)-(-)-phenylalanine. Again, D and L are related to which enantiomer of a compound is being discussed. Less obvious, is how D and L are derived. Without going into detail, it is necessary to compare the structure of the molecule being named to a molecule called glyceraldehyde. This is just the first of the problems with this naming system. The second is that it cannot handle diastereomers and has to give each one a completely different name. Just search the intertubes for the 16 stereoisomers of an aldohexoses (simple sugars like glucose) if you want to see how confusing it can get.

D & L enantiomers and their connection to glyceraldehyde
Systematic determination of stereochemical descriptors
The systematic naming now used, employs the descriptors R and S to define each stereocentre within a molecule. This is known as the Cahn-Ingold-Prelog (CIP) nomenclature. The steps to determine the stereochemical descriptor of a stereocentre are outlined below:
Identify the stereocentre.
Rank the substituents (groups) attached to the stereocentre. To rank the groups follow these rules:
Rank the four atoms directly attached to the stereocentre in order of decreasing atomic number. Highest atomic number is given the highest priority.
If any atoms directly attached to the stereocentre are identical then look at the three atoms attached (ignore the stereocentre). If there is now a difference, the atom with the highest priority atom attached is ranked next. If there is no difference move to the next atom.
A double or triple bond counts as two or three bonds to the same atom.
Keep moving along a substituent until you find a difference. If there is no difference you are not looking at a stereocentre!
- Orientate the molecule so that the lowest ranked group (priority 4) is pointing away from you. Be careful: it is easy to swap the position of groups around at this point and accidentally draw the wrong stereoisomer.
- The lowest rank group is now ignored. Draw an arrow linking rank 1 to rank 2 to rank 3. If the arrow is clockwise (to the right) the stereocentre has R configuration. If the arrow is anti-clockwise the stereocentre has S configuration.
You must ensure that the lowest priority group points away from you
Example
An example might make it clearer. What is the stereochemical descriptor of the enantiomer of carvone drawn below?

Working through an example to determine the stereochemical descriptor of carvone
The first step is to determine the stereocentre. Here is the carbon with four different groups coming off it at the bottom of the molecule. Neither alkene is a stereocentre. It is impossible to change the geometry of the internal alkene (the one in the ring). Swapping the orientation of the carbon marked with a 3 would prevent the ring from forming, the atoms would be too far a part to link together. The other external alkene has two identical substituents (hydrogen atoms) on one carbon so is not a stereocentre.
In the second step you rank the substituents of the stereocentre. First, rank the atoms attached directly to the stereocentre according to their atomic number. This is 1st atom in diagram above. It is easy to spot the lowest ranked group, it is the hydrogen atom (atomic number = 1), and this is ranked 4. The other three groups all have a carbon atom attached to the stereocentre. To differentiate between them, move to the next atoms along each chain or what I have called 2nd atom. Here the alkene hanging off the ring has three carbon atoms compared to the other groups that only have a single carbon atom. So it has the highest rank, 1. The other two groups are the same at the second atom (both are CH2. Moving to the next atoms along the chain, 3rd atom, permits differentiation. One has an oxygen atom (ranked 2) and the other just carbon (rank 3).
Having ranked each group on the stereocentre, you check the oritentation of the lowest priority group. It must point away from you. In this example, that means flipping the molecule through 180°. Once the lowest ranked group is pointing away it can be ignored. Finally, draw a line connecting the ranked atoms 1 to 2 to 3. If the line is anticlockwise, as shown, the molecule is S. If the line was clockwise (to the right) it would be R.
Alkene nomenclature
The geometry of an alkene can also be given a stereochemical descriptor. This is based on the same priorities as the CIP rules above. The two possible descriptors are:
E-alkenes which have the two highest priority groups on opposite sides (trans)
Z-alkenes, which have the two highest priority groups on the same side (cis)
To determine the stereochemical descriptor of an alkene:
Divide the double bond in half.

Consider each carbon atom separately
- For each end determine the highest priority substituent. To rank the groups use the CIP guidelines. These are summaries for a second time below (practice makes perfect):
- Rank the atoms directly attached to the double bond carbons. Highest atomic number is given the highest priority.

Ranking substituents of an alkene
- continued
- If the atoms directly attached are the same, move to the second, third, fourth atoms away until a difference is found
- Multiple bonds have the same number of single-bonded atoms (a double bond counts as two single bonds to the same atom)
- If the two highest priority groups are on opposite sides of the double bond it is described as E. If they are on the same side, the stereochemical descriptor is Z.
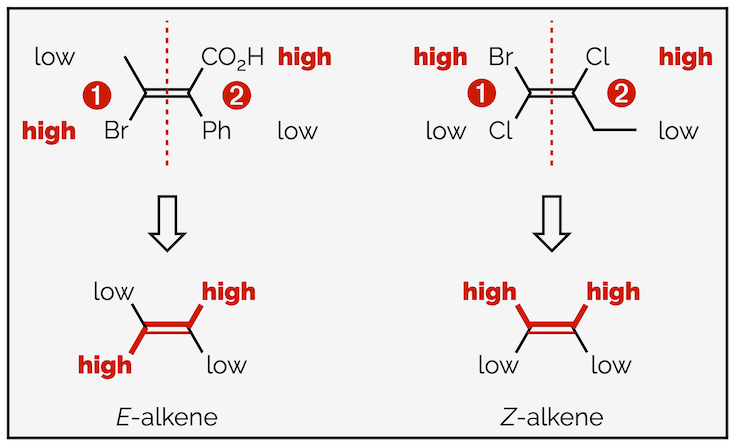
Alkene stereochemical descriptors
The Conclusion (to naming but not this post)
This has just been a brief introduction to determining the stereochemical descriptors for stereocentres (tetrahedral) and alkenes. There are guidelines for applying these descriptors to stereogenic axis and planes. Helical structures use different descriptors. The CIP rules can get more complex, there are rules governing any difference that is possible in a chain (for example A Z-alkene has higher priority than an E-alkene (and even this is a simplification)). But very few people understand or apply these rules confidently. Luckily computer apps can give provide names if the molecules are drawn well.
Drawing Tricks
Stereochemistry isn’t difficult if you are careful (fateful last words … there are probably mistakes in this post). Most mistakes are made when re-drawing or manipulating representations of stereoisomers, for example when rotating a molecule to orientate the lowest priority away from you. Below are a couple of tricks that we find useful when drawing molecules with (tetrahedral) stereocentres. (Of course, the first trick is not to rotate a molecule if the priority 4 substituent is pointing in the wrong direction … just remember to reverse your answer!)
The first shows the two ways to draw an enantiomer. These only differ by where you place the mirror that reflects the molecule.
In the first drawing, the mirror has been placed to the side of the original structure. To draw the molecule reverse the order of the bonds left-to-right but keep the wedges and hashed lines the same. This is easiest mirror image to visualize but, in my experience marking exam scripts, causes the most issues in terms of drawing the molecule accurately.
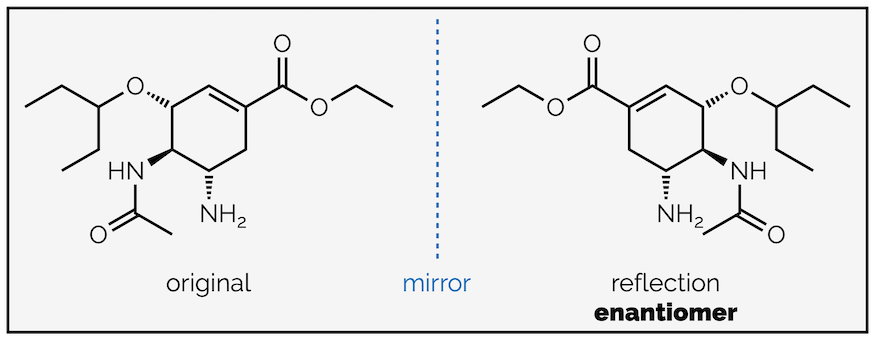
Enantiomers with the mirror to the right of the original molecule
The second way to draw an enantiomer is reflect the molecule in the plane of the page/screen. This means the mirror is directly below the molecule. Students frequently struggle to visualize this but it makes drawing the enantiomer easier. All atoms stay in exactly the same place and all you have to do is change the bold wedges to hashes and vice-versa. Two attempts to draw this are below. The top literally has the mirror in the plane of the screen while the bottom tries to show this process with forced perspective:

Enantiomers reflected in the screen
or …

Enantiomers reflected in the screen (right) and forced perspective drawing of this process (left)
The second trick involves the manipulation of stereocentres in drawings. This allows you to rapidly rotate bonds. The key is to remember that our skeletal drawings of stereocentres are a two-dimensional representation of a three-dimensional shape. For a tetrahedron, a maximum of two bonds (lines) can be in the plane of the page. The other two must be either up (outwards) or down (into the plane). In a drawing, two bonds will be normal lines in the plane of the paper while there should be one upwards wedge and one downward hashed line. So far, nothing new.
Commonly, your drawing will show just three bonds of a stereocentre, with the fourth, to a hydrogen atom, ignored in skeletal representations. In this drawing only two bonds can be the plane of the page, the other bond will either be a wedge or a hashed line. In the example below it is a bold wedge. And here is the trick … as long as you keep the position of all the atoms the same then any one of them can be the wedge. Each drawing shows the same configuration. This effectively shows you rocking the tetrahedron back and forth (about 54°) but not fully rotating it.
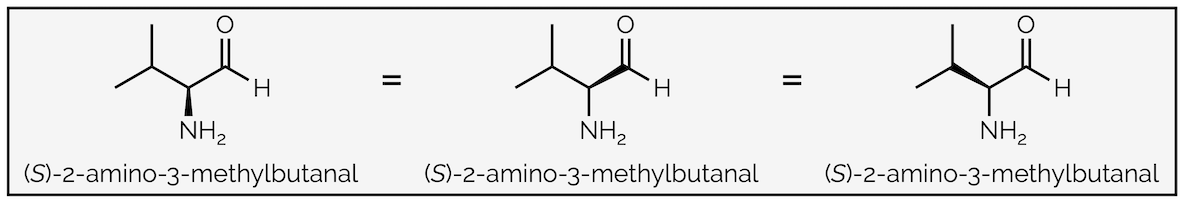
‘Wobbling’ a bond - if you keep the atoms in the same place the stereocentre remains a bold wedge
The final trick allows you to rotate a bond fully. Taking the same drawing it is possible to fully rotate around a bond if you swap two atoms. When you do this you must change a bold wedge into a hash or vice versa.

Rotating a bond by swapping the position of the groups and reversing wedge/hash
I recommend that you make a model (easily achieved with wine gums and toothpicks...also tasty to eat afterwards) and prove to yourself that these shortcuts work.
Conclusion
Naming is not fun. But it is (a) sometimes necessary, and (b) sometimes very useful to check that you haven’t made any mistakes. Here I have introduced how to assign stereochemical descriptors to tetrahedral stereocentres and alkenes. Most mistakes get made when manipulating the drawings so the last section adds a couple of cheats for easily re-drawing compounds.
As always, the best way to improve your skills is by practice so here are some QUESTIONS (and answers).