Asymmetric Hydroboration
Introduction
Hydroboration-oxidation is a useful method to transform alkenes into alcohols. The reaction is regioselective, giving the anti-Markovnikov product, and is also stereospecific with the boron and hydrogen adding to the same face of the alkene (a concerted syn-addition). The oxidation of the organoborane (C–B bond) occurs with retention of stereochemistry so that the alcohol will be on the same face as the hydrogen atom.

An example of hydroboration-oxidation. The reaction is regioselective and stereospecific.
Anti-Markovnikov addition is shorthand for the boron (heteroatom) adding to the least substituted carbon of the alkene (see HERE). This selectivity can be explained by both steric and electronic arguments. For this summary, it is the steric argument that is most important. The boron adds to the least substituted end of the alkene because it is larger than a hydrogen atom and minimization of repulsion places it further from any sterically demanding groups on the alkene. Use of a bulky organoborane (R2BH) instead of borane (BH3) increases the regioselectivity as there is greater steric interactions.

The transition state for hydroboration. For steric reasons, the boron adds to the least hindered end of the alkene. This avoids unfavorable repulsive interactions.
The addition is stereospecific as it is a concerted processes with all the bonds being made and broken at roughly the same time. This means the boron and the hydrogen must add to the same face of the alkene in an example of a syn-addition.
The steric interactions don't just effect which end of the alkene the boron adds (regioselectivity), they can influence which face of the alkene reacts, and this leads to stereoselectivity. Hydroboration, and the subsequent oxidation, can lead to either diastereoselective addition if there is an existing stereocenter in the alkene or enantioselective addition if the organoborane contains the stereocenter.
In this summary, I will discuss substrate control, where the alkene controls which face is attacked, and reagent control, where the organoborane controls which face of the alkene is attacked. I’m not going to cover catalyst control. There are many elegant catalytic systems that can control selectivity (rhodium was popular when I was a young chemist) but here I just want to introduce basic concepts without having to worry about a change in reaction mechanism. Those that are interested in taking this further and looking at catalyss coould do a lot worse than starting with a review such as Chem. Soc. Rev., 2022, 51, 8877-8922.
Hydroboration - Substrate Control (Cyclic)
The minimization of repulsive steric interactions results in addition occurring to the least hindered face of the alkene. This is readily visualized in the reaction of cyclic molecules. The classic example is the hydroboration of pinene.

The borane adds to the least hindered face of pinene. This is opposite the two methyl groups (shaded in grey), which block approach to the top of the alkene.
The bridge across the ring prevents the six-membered ring from adopting the common chair-like conformation. The model of pinene below is based on X-ray crystallography data (Acta Cryst. 'E57' (11): o1039-o1040. DOI:10.1107/S1600536801016415), while the model of alcohol was created in ChemDoodle3D but closely resembles this fragment in larger x-ray structures.

Representations of the three-dimensional structure of pinene and the alcohol that is formed on hydroboration-oxidation ((+)-Isopinocampheol).
The hydroboration (without the oxidation) of pinene is a useful example for two reasons. Firstly, it clearly shows the addition of borane occurring to the least hindered face. One of the bridgehead methyl groups is fixed above the alkene and blocks approach to the top face (as I've drawn it) of the alkene. Secondly, it gives a new organoborane is a useful chiral reagent that will crop up later.
Conformation of Acyclic Alkenes with Allylic Stereocenter
It is possible to obtain excellent substrate control with acyclic systems as well but the molecules have more conformational freedom so it can be harder to predict. Most undergraduates are introduced to the conformational analysis of simple alkanes (ethane, propane and butane) as well as cyclohexane (with its lovely chair conformation). Many don't get to see the effect of an alkene so I will start by looking at the conformations of propene and introducing allylic strain or A-strain. The two extreme conformations are shown below:

The two extreme conformations of propene. The favored conformation minimises A1,2 strain but at the expense of A1,3 strain.
The barrier to rotation in propene is only 8.4 kJ mol–1. This is much less than the barrier in propane (14.2 kJ mol–1), meaning rotation is easier. The bonds in propene are further apart than in propane with bond angles of 120° compared to 109°. The barrier to rotation in propene is largely the result of electronic interactions but as soon as there is an allyl substituent (one of the hydrogen atoms is replaced by anything else) steric interactions are more important.
The four important conformations of but-1-ene are shown below. Again, the most stable or favorable conformation has a hydrogen atom eclipsing the alkene. This is known as A1,3 eclipsed. This is called the skewed conformation.

The main conformations of but-1-ene. The most stable conformation has the smallest allylic substituent eclipsing the hydrogen at the 3-position. The least favored conformations suffer from A1,2 strain with these substituents being eclipsed. It also shows that A1,3 strain can be important. Barriers to rotation are relative to 0 kJ mol^–1 for the most stable conformation and are taken from J. Am. Chem. Soc. 1985, 107, 5035.
The bisecting (A1,3 staggered) conformations are disfavored due to A1,2 strain (A1,2 eclipsed). Both have a similar barriers to rotation but the methyl group causes greater steric strain so the bisecting trans conformation is slightly higher in energy. But again, the barriers to rotation are low compared to simple alkanes. This means that allyl systems with such substitution patterns tend to give poor or unpredictable substrate control (unless there is a stereoelectronic effect such as a silyl substituent but that is a whole different summary).
Larger molecules, such as pent-2-ene, that have a cis alkene have more well-defined conformations and prediction of stereochemical addition becomes more reliable. Once the alkene is disubstituted, there can be a larger barrier to rotation. There are two important situations. The first involves cis or Z-alkenes. These lead to substantial A1,3 strain. The second system requires a substituent at the 2-position, which leads to A1,2 strain.
The important conformations of Z-pent-2-ene are shown below:

The important conformations of Z-pent-2-ene. The relative energies are taken from David Evans' Chem 206 lectures (found HERE), with the exception of the gauche conformation, which is an approximation. The key interactions are now the A^1,3 strain between the allylic position and the methyl group on the alkene.
The most stable conformation has an eclipsed hydrogen as this minimizes steric interactions A1,3 with the methyl group. This conformation is also favored by hyperconjugation with the C–C σ bond of the allylic position overlapping the π* antibonding orbital of the alkene resulting in a stabilizing delocalization of the electrons (J. A. Chem. Soc. 1987, 109, 6591 & J. Am. Chem. Soc. 1985, 107, 5035). It has been argued that the largest substituent on the allylic position (methyl group in this example) will be perpendicular to the alkene in the most stable conformation. This latter conformation certainly might be important in the reaction transition state as delocalization into the π bond would increase the nucleophilicity of the alkene. Ultimately, it doesn't matter as there appears very little energy difference between this and the eclipsed conformation (Chem. Rev. 1989, 89, 1841).

Favored conformation of Z-pent-2-ene.
There are two staggered or bisecting conformers. These are disfavored due to A1,2 strain, where either the methyl group or a hydrogen eclipse the hydrogen at the 2-position of the alkene.
I cannot find values for both conformations and the literature implies that the trans conformation is energetically disfavored due to A1,2 strain being greater than the steric interaction of the two methyl groups. This seems odd considering the methyl groups must be at similar distances to the syn-pentane interaction.
It doesn't matter as the barrier to rotation in this system comes from the second A1,3 eclipsed conformation. This is the least stable conformation and has the maximum interaction between the methyl groups. This causes a greater barrier to rotation than the syn-pentane interaction (16.2 kJ mol–1 for A1,3 versus 15.4 kJ mol–1 for the syn-pentane interaction).
The second important allylic system has a substituent at the 2-position and results in A1,2 interactions controlling the barrier to rotation.

A^1,2 strain controls the barrier to rotation in 2-methylbut-1-ene.
Once again, the most stable conformation sees the small hydrogen atom of the allylic position eclipsing the alkene. This minimizes A1,3 strain and has the substituents of the allylic position gauche to the methyl at the 2-position. This minimizes A1,2 strain.
The least stable conformation or the barrier to rotation, is a bisecting conformation with the A1,2 methyl groups eclipsing. This results in the maximum torsional and steric interactions. The other two conformations are not as important as they have a small hydrogen atom interacting with the methyl. Neither is as important as the repulsion caused by two methyl groups.
A1,3 strain is more destabilizing than A1,2 strain, but in complex alkenes the favored conformation will try to minimize both.
Hydroboration - Substrate Control (Acyclic - allylic stereocenter)
Hydroboration of an acyclic alkene with an allylic stereocenter can occur with high diastereoselectivity. A classic example comes from Kishi's synthesis of monesin (J. Am. Chem. Soc. 1979, 101, 259). The scheme shows that the reaction occurs with 92:8 diastereomeric ratio or d.r. This means 92% of the product has the configuration drawn, with only 8% as a different diastereomer (I have normalized to a total of 100 as I think it is more descriptive than the ratio given in the paper, which was 12:1).

The hydroboration-oxidation of an intermediate in the synthesis of monesin. The three most important conformations are shown as Newman projections.
The major diastereoisomer is formed through a transition state that minimizes A^1^,^3 strain. The conformation of the substrate in the transition state is probably staggered rather than eclipsed (closer to the conformation that has the largest substituent perpendicular to the alkene). Having the large substituent anti to the approaching borane increases nucleophilicity of the alkene through hyper conjugation.
Of the two conformations that place the large group perpendicular to the alkene, the one that minimizes A1,3 strain is favored. This places the hydrogen atom almost eclipsing the alkene in what is called the inside position; the hydrogen is between the alkene and the borane. This favors approach of the borane from the bottom face as shown. The diagram shows both the transition state as a Newman projection and the resulting product of hydroboration as a Newman projection. I have then unfolded the Newman projection and added the stereochemical descriptors to the diagram (assuming that the borane has been oxidized. A quirk of the CIP stereochemical descriptors is that the stereocentre changes from R to S on oxidation due to the O and B changing priorities as they are either side of carbon on the periodic table).
The other conformation with the large group anti to the borane has a destabilizing A1,3 interaction between the methyl group and the CH2OH of the alkene.
The minor diastereomer probably arises from the other conformation that minimizes A1,3 by placing the hydrogen in the inside position. This would have the methyl group anti to the borane. It is disfavored due to the repulsion between the borane and the largest substituent.
The preferred conformation can be summarized as shown in the diagram below:

A summary of A1^,3 strain controlling the diastereoselectivity of hydroboration of an alkene with an allylic stereocenter.
The hydroboration of 1,1-disubstituted alkenes shows both substrate control and reagent control with small boranes giving one stereoisomer and larger organoboranes giving the opposite diastereomer. An example of this comes from the synthesis of ionomycin (J. Am. Chem. Soc. 1995, 117, 3448). The small borane dimethyl sulfide complex leads to the syn diastereomer while using the bulky reagent, 9-BBN, gives the anti-diastereomer.

Diastereoselective hydroboration-oxidation of ionomycin is an example of A^1,2 control and reagent control.
The change in selectivity can be understood if you look at the Newman projection of the most important conformations. I'll start with the reaction of the small borane first. This is controlled by the minimization of A1,2 strain. The boron adds to the unsubstituted end of the alkene (anti-Markovnikov addition) and anti to the largest group on the stereocentre. This leads to to two possible conformations. The favored conformation has the hydrogen in the outside position to minimize A1,2 strain. There is an unfavorable interaction between the borane and the alcohol but as the reagent is small this can be ignored. The alternative, disfavored, conformer has destabilizing A1,2 strain as the CH3⟷OH interact.

A^1,2 strain controls the reaction of a 1,1-disubstituted alkene and a small borane. The disfavored conformation has an interaction between the larger hydroxyl group and the methyl group. The favored conformation places the hydrogen outside, effectively eclipsing the methyl group. The hydrogen is small and this interaction is unimportant. This does place the hydroxyl group inside but the small size of the borane means this is acceptable.
When a large organoborane (RB ≠ H) reagent is used the situation reverses. Now the interaction between the reagent, and the inside substituent becomes important, to the point that it can override A1,2 strain. So, with big reagents, the small hydrogen in the inside position is favored.

When a larger organoborane is used A^1,2 strain is not the most important interaction and now the interaction between the inside group and the reagent becomes important.
The diagram above shows that the interaction between the organoborane and the inside substituent on the allyl stereocenter controls the selectivity. The disfavored conformation has minimal A1,2 strain but maximizes this new steric repulsion (reagent & medium substituent of allylic stereocentre). The favored conformation places the smallest substituent in the inside position at the expense of A1,2 strain. The diagram shows this interaction in both a forced perspective representation and a Newman projection. It then unfolds both representations to show how you get back to the more familiar skeletal representation.
The hydroboration of a 1,1-disubstituted alkene can be summarized in the diagram below:

A summary of the effect of changing organoborane on the hydroboration of 1,1-disubstituted alkenes with an allylic stereocenter.
Hydroboration - Reagent Control
The first example of substrate control that I presented was the hydroboration of α-pinene. Reaction of pinene with an excess of borane results in hydroboration occuring twice. The bulk of the bicyclic ring hinders the third addition from occurring. The resulting dialkylorganoborane is called diisopinocampheylborane or ipc2BH. The disproportionation of ipc2BH leads to a monosubstituted organoborane, monoisopinocampheyl borane ipcBH2. Both reagents have at least one B–H bond and have been used in the diastereoselective hydroboration-oxidation of achiral alkenes to give enantiomerically enriched alcohols.

The two classic chiral hydroboration reagents, ipc2BH and ipcBH2.
As an annoying aside, note how the direction of the optical rotation switches from positive to negative even though the stereocenters within each reagent remain the same. This shows the pitfalls of relying on old nomenclature when assigning a stereochemical descriptor to molecules. It is why it is useful for a synthetic chemist to have a working knowledge of the CIP-system for assigning stereochemical descriptors as outlined in an earlier summary (HERE).
The difference between substrate control and reagent control lies in which reactant contains a stereogenic element (or the controlling stereogenic element). In the examples above, the substrate had a stereocenter that influenced the approach of the reagent (borane). In reagent control, the situation is reversed, the reagent (organoborane) has one, or more stereocentres, and these control which face of the alkene it approaches. Reagent control allows you to synthesize either enantiomer (or diastereomer) at will (as long as both enantiomers of your reagent are available (which isn’t always the case)).
The example below demonstrates how reagent control can deliver each epimer at will. This example is taken from the synthesis of tylonolide (J. Am. Chem. Soc. 1982, 104, 5523). Each enantiomer of the organoborane leads to a different diastereomer with the same efficiency. This suggests that the other stereocentres of the substrate are sufficiently far away from the point of reaction that they have no influence.

Synthesis of two diastereomers of an alcohol. The choice of enantiomer of the organoborane determines which diastereomer is isolated.
The difference in size between the two reagents (the presence or absence of a second pinene moiety) means they are suited to reaction with different alkenes. IpcBH2 is smaller, with just one bicyclic substituent, it gives better results with bulky alkenes, trisubstituted alkenes and trans-alkenes. Ipc2BH is bulkier and gives good results with cis alkenes and 1,1-disubstituted alkenes.
To understand the diastereoselective addition of these reagents to an alkene it is best to start with ipcBH2 as you only need to consider the interaction of one isopinocampheyl unit with the substrate. IpcBH2 gives good results with trans-alkenes. If the ends of the alkene are different, the organoborane will add to the least sterically demanding end of the alkene and the hydrogen to the bulkier end. The organoborane could add with the ipc being syn or anti to the closest substituent. Clearly the anti-approach is favored as shown below.
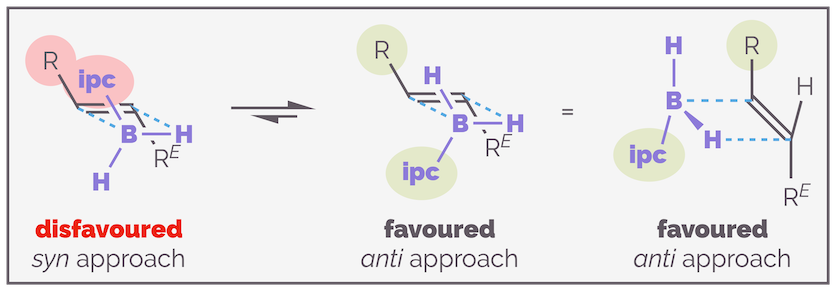
The smaller ipcBH2 reagent approaches a trans-alkene so that the boron adds to the least hindered end of the alkene and the isopinocampheyl unit is anti to other alkene substituent.
To determine the favored conformation of ipcBH2 as it nears the alkene, you can draw a Newman projection looking along the B–C bond; the hydrogen is the smallest substituent, while the methyl substituted half of the ring is the largest. I have drawn this below:

A Newman projection showing the relative sizes of the substituents on the stereocenter attached directly to the boron atom. This gives an idea of the steric environment around the reactive BH2 center.
Calculations by Houk suggest that when the organoborane adds to an alkene, it adopts a staggered transition state, minimizing torsional strain. The largest substituent will be anti to the new C–B bond as this leads to the greatest separation of reagent and substrate. The choice then becomes whether to place the hydrogen (small substituent S) in the inside position, close to the alkene substituent RE or if the medium (M) sized group is in this position (see below). Approach of the organoborane to the face of the alkene that leads to the former transition state is favored as it minimizes non-covalent interactions. If the organoborane adds to the opposite face of the alkene there is a disfavored interaction with the medium group and the substrate (as shown on the right-hand side of the diagram).
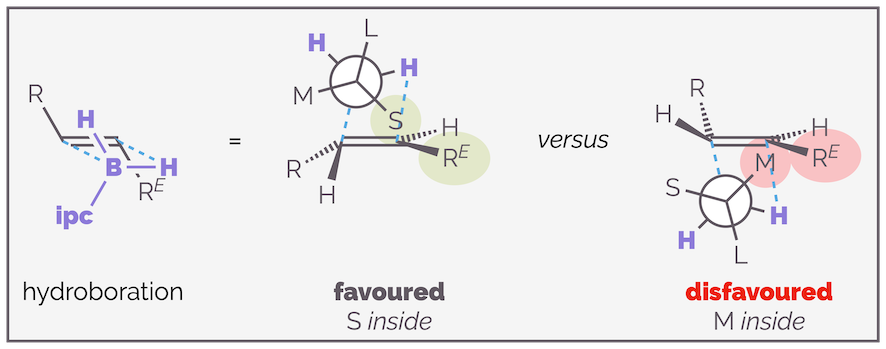
The preferred face of hydroboration is controlled by the interaction between the ipc moiety and the R^E substituent of the alkene substrate. The favored conformation places the least sterically demanding part of the organoborane (the hydrogen atom) close to the alkene.
The interaction between the isopinocampheyl of ipcBH2 and the substrate (RE) is essential for good stereoselectivity. The reaction of cis alkenes give poor results as this interaction is missing. Both alkene substituents are anti to the reagent, and without this interaction appraoch to either face is equally probable.

There is no selectivity with cis alkenes as the substituent is on the opposite side of the alkene to the reagent. If the reagent cannot interact with the alkene there is no communication of stereochemical information.
The reaction of ipc2BH is harder to visualize as you have to consider the conformation of two isopinocampheyl groups and how they interact with each other prior to the reagent approaching the alkene. Calculations by Houk (Chem. Rev. 1993, 93, 2439; Science 1996, 231, 1108; Tetrahedron 1984, 40, 2257) show that the transition state conformation for ipc2BH has the two isopinocampheyl groups in an eclipsed conformation with the small hydrogen atom overlapping the large side of the bicyclic ring. This leads to the two medium substituents being in a syn-pentane-like arrangement. In the immortal words of either Elvis Costello or David Byrne, ‘talking about music (conformations) is like dancing about architecture.’ In other words, I can't describe the confromation, you need a picture ...

This diagram attempts to show the conformation of ipc2BH in the transition state during addition to an alkene. This is not the ground state conformation of this molecule. I have tried to show that the C–H bond is eclipsing the large substituent of the isopinocampheyl ring, while the two medium sized substituents are eclipsed. One of the isopinocampheyl rings projects its bulk downwards while the other has the hydrogen in this position. This second ring behaves as if it had less steric bulk.
This conformation is unusual. The ground-state conformation for diisopinocampheyl borane minimizes syn-pentane-like interactions and has the two bicyclic groups staggered. If you are very lucky, I will discuss the favored conformation in a blog post about asymmetric allylations or aldol reactions.
This conformation of the reagent effectively differentiates the two isopinocampheyl rings. One of them will present the large, methyl substituted side of the ring to the approaching alkene while the other presents the hydrogen atom. This means one ring appears to be more sterically demanding than the other. In the diagram above, I have made the assumption that the alkene approaches from the bottom face. This makes the ring shown as a Newman projection smaller than the back ring.
When ipc2BH and an alkene react, one isopinocampheyl ring must eclipse an alkene substituent. Ideally, this will be the smaller isopinocampheyl ring to minimize steric repulsion. The approaches of different alkene geometries are shown below. This diagram explains why ipc2BH normally gives better results with Z-alkenes than E-alkenes. E-Alkenes always suffer from a steric interaction between the larger ring and the alkene. In Z-alkenes, the large ring can be anti to the substituents.

The approach of ipc2BH and an alkene minimizes steric interactions. The interaction of the large isopinocampheyl ring with the alkene is minimized. As a result, reaction with the Z-alkenes tend to be more efficient as this allows the large ring and the substituents on the alkene to be anti.
Once you have sorted out the conformation of the two isopinocampheyl rings and the relative orientation of the organoborane to the alkene you can now ignore the larger ring. All that is left to do (ha ha), is determine which face of the alkene is attacked. Luckily this is the same as with the previous reagent. The alkene will approach so that the small hydrogen atom on the small ring is in the inside position. This is shown for a Z-alkene below:

The enantioselectivity is determined by which face of the alkene the organoborane approaches. This is determined by the minimization of steric interactions. The substituents of the alkene will be close to the so-called small ring of organoborane. The alkene will attack so that the hydrogen atom of the isopinocampheyl ring is in the inside position.
Conclusion
Hydroboration-oxidation permits the anti-Markovnikov hydration of alkenes. It is a stereospecific reaction, with the hydrogen and boron (and the subsequent oxygen) undergoing syn addition through a concerted addition. The reaction can also be highly stereoselective. Steric factors influence which face of the alkene attacks the organoborane. This allows substrate controlled hydroboration, where a stereocenter within the reactant controls the interaction between reactants. It is also possible to move the stereocenters on to the reactant instead of the substrate. This permits reagent controlled hydroboration. This means the stereochemistry of the organoborane now controls which face of the alkene reacts. The two pinene-derived reagents discussed above are the classic examples of asymmetric reagent controlled hydroboration. They provide a wonderful framework with which to introduce many of the concepts we need to start rationalizing stereochemical induction. The same principles can be applied to other reactions (especially allylations and aldol reactions which can use other pinene-derived organoboranes). But, better, more reactive and/or more selective organoboranes have been developed over the last 30 years. This summary isn't meant to be review of asymmetric reagent controlled hydroboration so all I'll say is that you could do a lot worse than look at Angew. Chem. Int. Ed. 2009, 48, 1896 or Chem. Soc. Rev. 2022, 51, 8877.